 下载格隆汇APP
下载格隆汇APP
 下载诊股宝App
下载诊股宝App
 下载汇路演APP
下载汇路演APP
 社区
社区
 会员
会员
微信公众号:蹊之美股生物医药 / CaesarBiotech
ADCs药物肿瘤治疗综述全文共五块内容,分别为:
背景
1. 靶点-抗体的选择和优化
2. 连接子的设计和优化
3. 细胞毒药物的选择及研发公司
4. 新型的特异性位点偶联技术
其中,背景、靶点-抗体的选择和优化、连接子的设计和优化已在第一遍文章中介绍,此文为第二部分,主要是介绍ADCs药物的细胞毒药物的选择及研发公司、新型的特异性位点偶联技术。
第一部分内容见:
Antibody–Drug Conjugates ADCs:肿瘤治疗领域最有前景的方向之一(一)
3. 细胞毒药物
通常在细胞表面存在数量有限的抗原(每个细胞约5000-106 个抗原),大部分临床试验的ADC药物的DAR在3.5-4之间,最终进入细胞内的药物很有限,这就是为什么传统的化疗药(如甲氨蝶呤methotrexate, 紫杉烷类taxoids,蒽环类药物anthracyclines)不用来制作ADC药物的原因。目前大部分ADCs携带的细胞毒药物为靶向DNA和微管的药物。
微管抑制剂:maytansinoids-美登木素生物碱衍生物(如emtansine DM1,mertansine DM4);auristatins-奥瑞斯他汀衍生物(如mono-methyl auristatin E (MMAE), mono-methyl auristatin F (MMAF) )。这些药物通过对微管蛋白的解聚作用诱导细胞死亡。DNA损伤药物:插入DNA抑制拓扑异构酶2(topoisomerase 2)活性药物(如阿霉素 doxorubicin),裂解DNA的药物(如卡里奇霉素 calicheamicin ),烷基化DNA药物(如 DGN462),抑制DNA损伤修复有关的酶(拓扑异构酶抑制剂 SN38)。
疏水性的细胞毒药物容易导致抗体凝集,减少药物储存时间和在体内循环中的存留时间,而且这些药物被修饰后成为可连接时,是需要可溶性的,并在成为ADC药物后能保持在水溶质中的稳定性。当然还需要在生产时能以不错的成本来合成,最近被批准的Adcetris(brentuximab vedotin)和T-DM1(trastuzumab emtansine)它们携带的细胞毒药物分别为瑞奥斯他汀auristatin (Seattle Genetics technology)和美登木素生物碱maytansinoid (ImmunoGen technology),就在这些属性上非常的不错,而这两者细胞毒药物占到所有ADC药物研发的60%以上。其他的一些细胞毒药物包括:吡咯苯二氮平类药物(pyrrolobenzodiazepines PBDs), 吲哚苯二氮平类药物(indolinobenzodiazepines), 伊立替康衍生物(irinotecan derivatives), duocarmycins, 微管蛋白类(tubulysins)。由于存在激烈的竞争,还有不少尚未披露的细胞毒药物正在研发。
3.1 Auristatins-奥瑞斯他汀衍生物
这是ADC药物研发中最多的一类,包括MMAE和MMAF,并有两种不同的连接子。技术拥有者为西雅图基因公司Seattle Genetics,相关技术授权给AbbVie, Astellas/Agensys, Bayer, Celldex, Genmab, GlaxoSmithKline (GSK), Pfizer, Progenics, Roche/Genentech and Takeda/Millenium等公司。MMAE和MMAF均是尾海兔素10的类似物(dolastatin 10),这是一种天然的抗有丝分裂药物,从海兔(Dolabella auricularia)中提取,单药由于毒性太大,不能单独使用。MMAE和MMAF两者的优点是药物效能高、水溶性、生理状态下稳定、能与连接子稳定连接。还有其他的奥瑞斯他汀衍生物也在被其他公司研究(包括Ambrx, Bayer, Pfizer, Novartis, Pierre Fabre and Sanofi/Genzyme)。
3.2 maytansinoids-美登木素生物碱衍生物
这是ADC药物中第二大类研发最多的药物,主要包括DM1和DM4,并有4种不同的连接子。技术拥有者为ImmunoGen,相关技术授权给了Amgen, Bayer, Biotest, CytomX, Novartis, Roche/Genentech, Sanofi and Takeda等公司。DM1和DM4是美登木素的衍生物,是一种天然的benzoansamacrolide(苯并**大环内酯类),从非洲灌木Maytenus ovatus的树皮中提取,这类药物在微管中的绑定位点和长春碱类药物相同,但是具有更强的毒性。此药由于全身毒性太大,在作为单用抗肿瘤药物的临床试验失败。但在做为ADC药物的构建时,具有极好的稳定性和不错的水溶性。
3.3 苯二氮平类药物Benzodiazepines
PBDs是抗肿瘤的抗生素类药物,绑定至DNA序列特异性小凹槽,此药是Spirogen研发的(现在属于阿斯利康),授权给了多个公司,如Seattle Genetics, Roche/Genentech, Stemcentrx (now part of AbbVie), ADC Therapeutics, Kolltan Pharmaceuticals and Mitsubishi Tanabe Pharma。两个PBD单位采用不同系链聚合而产生对称和不对称的聚合物,可以与DNA的鸟氨酸N2位点交叉结合,PBD的抗肿瘤作用是皮摩尔(picomolar)级别的。而且这药不是MDR1的底物,机制上可以针对此类耐药的肿瘤。自从2013年起,有10个以上携带PBDs的ADCs药物进入临床,这成为继奥瑞斯他汀auristatins和美登木素生物碱maytansinoids最有前景的负载药物。
5个PBD为基础的ADCs药物(如vadastuximab talirine discontinued),SGN-CD33A、SGN-CD70A、SGN-CD19B、SGN-CD123A和SGN-352A,由西雅图基因公司研发,分别针对AML (phase III), RCC (phase I), B-NHL (phase I), AML (phase I) 和 multiple myeloma (phase I)这5个指征,这里除了肾癌,其余均是血液病肿瘤。这几个都是最早公开的位点特异性的、同源性、第三代ADCs药物,抗体带有修饰过的半胱氨酸残基,靶点分别为CD33、CD70、CD19、CD123、CD352,连接子为蛋白酶可分解的缬氨酸-丙氨酸(valine-alanine)连接子,连接子通过SGD1882 PBD的苯胺与PBD相连接,DAR为2。
ImmunoGen公司在研发自己的苯二氮平类药物负载药物IGNs,公司的IMGN779用的PBD是DGN462,靶点为CD33,由于没有DNA交叉连接的特性(交叉连接能导致延迟的系统毒性),在有效性和耐受性上起到了平衡作用,目前进展到临床I期,适应症为AML。
还有其他PBD为基础的ADC药物,如AbbVie (Stemcentrx)的rovalpituzumab tesirine (Rova-T; also known as SC16LD6.5),是生物标记物的ADC药物,靶向肿瘤干细胞表面的Delta-like protein 3 (DLL3),包含了SG3199的PBD,DAR为2,连接子为可分解的缬氨酸-丙氨酸马来酰亚胺类型的连接子,适应症为非小细胞性肺癌,目前为临床III期,还有他们公司的SC-002 和 SC-003,也是PBD为基础的ADC药物,进入临床I期,适应症为SCLC和卵巢癌,靶点未公布。还有ADC Therapeutics公司的ADCT-301和ADCT-402,靶点为CD25和CD19,用的都是PBD的SG3249类型。
3.4 喜树碱类似物(Camptothecin analogues)
SN-38和DX-8951f分别被Immunomedics 和 Daiichi Sankyo公司用来制作ADCs药物,并已进入临床。
SN-38为抗肿瘤前药伊立替康irinotecan的活性代谢物,抑制DNA拓扑异构酶1(DNA topoisomerase 1 TOP1), SN-38由于毒性太大以及较差的水溶性,不能单独用于治疗肿瘤。Immunomedics公司的两个Antibody–SN-38 ADC药物为labetuzumab govitecan (also known as IMMU-130),以及sacituzumab govitecan (also known as IMMU-132; formerly known as isactuzumab govitecan), 均为水溶性,被设计成接近同质性,DAR为8,靶点分别为carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5),以及TROP2 (also known as TACSTD2)。其中TROP-2是一种细胞表面糖蛋白,在多种腺癌中有过表达。Labetuzumab govitecan进展到临床II期,sacituzumab govitecan进展至临床III期,指征为TNBC 3线用药,并获得美国FDA突破性治疗设计。虽然指征为3线用药,但临床二期出来的数据非常不错,根据临床II的数据,公司已在2018年上半年申请上市,同时III期临床试验在今年3月份已开始。
DX-8951f是水溶性的喜树碱类似物,它能有效控制MDR1介导的肿瘤多重耐药细胞,它的ADC药物为DS-8201a (developed by Daiichi Sankyo)靶点为HER2,连接子为maleimide–Gly-Gly-Phe-Gly肽链。在今年ASCO上公布了临床数据,34例Her2低表达的转移性乳腺癌中,50%的ORR,85.3%的DCR;在Her2-positive组的数据中54.5%的ORR及93.9%的DCR;效果不错。Grade 3的AE大于10%,而且有近9.5%的患者因安全性问题终止临床试验,Grade5(死亡)的患者有4%。由此看来,毒性还是比较大的。
3.5 Tubulysins
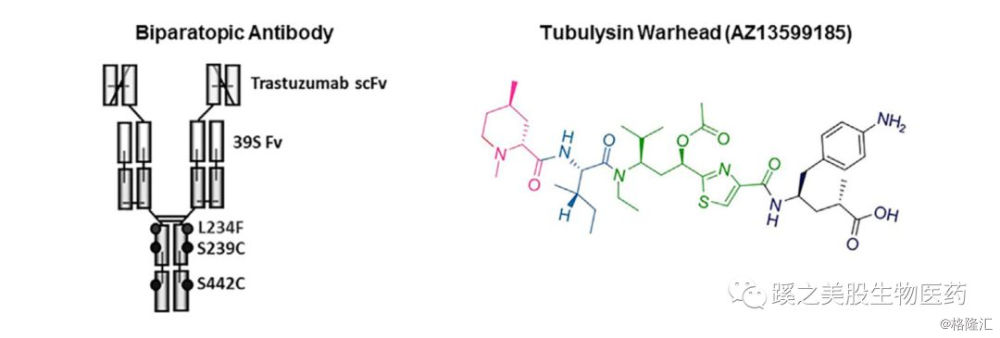
在2000年有Hofle等在粘细菌中提取出来,具有抗细胞有丝分裂活性的一类家族天然产物,细胞毒性机制和长春碱类似,能有效抑制微管蛋白聚合,干扰增殖细胞纺锤体形成,使细胞分裂停止在G2/M期,导致细胞凋亡。抗癌活性是(长春碱、紫杉醇等)的10-1000倍以上,还有抑制血管新生的作用。同时还有可能绕过DM1外排泵的作用,因此可以针对T-DM1抵抗的细胞。目前在研发的是AZ13599185 (developed by AstraZeneca/MedImmune),AZ13599185被连接到4个修饰过的半胱氨酸上,抗体为双特异性抗体,靶向HER2的两个非交叉的表位。ADC药物为MEDI4276,管线进展到临床I期,由于是交叉靶向1个抗原的两个表位,抗体比较容易在细胞表面形成簇,可以导致快速的内吞,强化细胞内溶酶体内的转运,甚至在低表达的HER2中也有杀伤作用。
MEDI4276(developed by AstraZeneca/MedImmune)的部分1期临床数据在2018年3月公布,这一管线的适应症为复发难治性晚期乳腺癌和胃癌,是计量爬坡试验。MEDI4276抗体的一个表位是HER2的39S,还有一个ECD4(即trastuzumab的靶点表位),它的细胞毒药物AZ13599185是Tubulysin的衍生物,双特异性抗体在Fc区域有3个位点被突变,分别是L234F、 S239C、S442C,每个重链上有2个修饰过的半胱氨酸残基(S239C and S442C),能特异性偶联(site-specic conjugation)连接子马来酰亚胺基己酸(maleimidocaproyl),DAR为4。位点L234F的突变能降低Fc gamma receptor (FcgR)的绑定,这一突变能最小化FcgR介导的不依赖HER2的正常组织的摄取,能降低诸如血小板降低的脱靶效应。结构图见下。
3.6 阿霉素类(Doxorubicin)
阿霉素类是放射菌类来源的抗有丝分裂抗肿瘤药,最早的ADC药物是BMS-182248 (also known as SGN-15; developed by BMS and Seattle Genetics),靶点为Lewis Y抗原,该抗原在多个肿瘤中表达,临床试验进入II后,数据也不错,但西雅图基因公司由于种种原因,停止了此管线的进展。Immunomedics公司的Milatuzumab doxorubicin (also known as IMMU-115) ,靶点为CD74,存在于血液肿瘤细胞的表面,进展至临床II期,适应症为慢性淋巴瘤(chronic lymphocytic leukaemia CLL) 和 非霍奇金淋巴瘤NHL。连接子为pH敏感性,在与CD74结合后,被内吞入细胞,CD47抗体再从新被转运至细胞表面,这样可以结合更多的ADC药物,没看见过这么傻的肿瘤。
3.7 Calicheamicins卡奇霉素类
这是一类高能效的烯二炔抗肿瘤药,是从放射菌类(Micromonospora echinospora)分离出来的,它能绑定DNA小凹槽,有位点特异性的分解双链DNA。卡奇霉素疏水性强,每个抗体只能携带较少分子。辉瑞的包含这个药物的ADC药物PF-06647263 已至临床I期,靶点为ephrin A4,用来治疗TNBC。
3.8 Duocarmycins
这是一类DNA小凹槽烷基化的药物,由Synthon研发的ADC药物SYD985(trastuzumab duocarmazine)已进入临床I期,在针对乳腺癌和胃癌模型上的数据显示,它能针对低表达的HER2的肿瘤细胞,有可能是相对T-DM1的“Biobetter”版本的药物。
3.9 其他
其他负载药物α-鹅膏蕈碱(α-amanitin an RNA polymerase II inhibitor),cryptophycins(效能优于MMAE和DM1),蒽环类药物anthracyclines,根霉素(rhizoxin也是一种微管抑制剂),spliceostatins 和 thailanstatins(两者均为RNA剪切体抑制剂)。
4. 新型的特异性位点偶联技术
二代ADC药物负载的药物具有不均一性(DAR在0-8之间),平均DAR为3.5或4。未偶联部分抗体通常没有活性,并且和药物负载的抗体竞争绑定抗原,这在裸抗体比例较少时对效果影响不大(比如brentuximab vedotin 和 trastuzumab emtansine他们的裸抗体在5%左右),但对于gemtuzumab ozogamicin含有50%的裸抗体,效果就会大打折扣。而且,DAR大于4的药物,通常耐受性低,更高的血浆清除率,在体内试验中效果也较低。
大部分ADC管线的药物有着共同的结构特点,比如硫化琥珀酰亚胺(thiosuccinimide)连接,这是硫醇和烷基马来酰亚胺的反应合成。这种类型的化学广泛使用,因为在生理状态下马来酰亚胺和硫醇能快速反应,而且产量很高。但是这个连接在生理状态下能缓慢可逆的分解。将近2/3的ADC药物都含有烷基化的马来酰亚胺,这样会导致药物在循环中的缓慢损失和脱靶效应。这在半胱氨酸连接的和赖氨酸通过硫醚连接子SMCC连接的ADCs药物中可见。这儿存在的问题可以通过特异位点偶联(site-specific conjugation)和其他偶联化学得到解决。
特异性位点偶联(site-specific conjugation)技术在第三代ADC药物中发挥重要作用,它能使ADC药物更好的控制DAR的比例,药物更加有钧一性,使ADC药物有更高的治疗窗。而且,这些ADC药物能连接更好的细胞毒药物,整合更好更新的抗体结构,比如说使用双特异性抗体。下面介绍几种比较前沿的特异性位点偶联技术,这些技术是否具有实用性,最终还需临床验证,但是,一旦验证成功,对于第三代ADCs药物的研发进展起极大的推动作用。
4.1 经侧链半胱氨酸残基法(Engineered cysteines)
在IgG抗体中引入特异性位点的半胱氨酸有着不同的水溶性和电荷,这种技术已被从多公司研发(比如Genentech, Seattle Genetics, Novartis, MedImmune, Kirin and Pfizer)。这种技术叫TDCs(THIOMAB drug conjugates TDCs),TDCs药物在循环中也存在较高的解偶联率,这是由于马来酸亚胺(maleimide)可以与血浆中的白蛋白、自由半胱氨酸、谷胱甘肽发生硫醇基团的交换反应。这个问题在下一代TDCs药物的研发中得到解决(通过轻链上V205C的突变),这个特异性位点选择在阳性电荷环境中时,可以导致琥珀酰亚胺的环被水解,从而防止硫醇基团的交换反应。
Vadastuximab talirine (also known as SGN‐CD33A; developed by Seattle Genetics discontinued )是在重链的S239C上加上了半胱氨酸,通过和可分解蛋白水解的连接子(valine-alanine)与PBD聚合物连接,接近同质,DAR为2,这个首个公开的管线进入临床试验的位点特异性联合技术。西雅图基因的SGN-CD70A和SGN-C19B也采用了相同的技术,两个药物都已进入临床试验,同样C-端硒代半胱氨酸也可以作为位点特异性连接。
4.2 非天然氨基酸修饰技术(Unnatural amino acid engineering)
基因编辑unnatural amino acids (UAAs)生物正交化学反应被用来制造位点特异性ADCs。通过改造tRNA合成酶去识别UAAs,从而在抗体上引入UAAs做为特异性位点。举个例子,auristatins可以连接至2个UAA肽上,而不一定要结合在20个经典的氨基酸肽链上,抗体上表达对位十二苯基丙氨酸(para-acetylphenylalanine pAcPhe) ,这种抗体能在哺乳细胞上表达,和野生型抗体在属性上很相近,优化肟连接反应使auristatin分子连接在pAcPhe上,auristatins连接至抗体的2个UAA上,这个化学反应高效,可控,生成的连接稳定,使ADC药物减少脱靶效应。
一种无细胞蛋白表达系统(cell-free protein expression system,eveloped by Sutro Biopharma )能加入UAA para-azidomethyl-l-phenylalanine (pAMF)至抗体的特异性位点,然后通过copper-free click chemistry (这是一种生物正交化学反应,叠氮-炔”链接"反应,可以有环加成作用,这种方法没有铜催化剂,因此没有细胞毒性)可以几乎完全的耦合连接dibenzocyclooctyne – PEG–MMAF。
叠氮化物抗体(AzAbs; developed by Allozyme)有特异位点修饰的叠氮化化学键,每个叠氮的位置是通过特异位点置入UAA实现的,这个技术的一个关键点是化学反应仅出现在叠氮位点,在抗体的其他位点上均无出现,因此,在完成生物正交连接后,连接子非常稳定,稳定性是马来酸亚胺或者硫酯连接的10倍。
4.3 酶辅助的连接技术(Enzyme-assisted ligation)
位点特异性化学蛋白连接也可以通过基因编码的氨基酸标记来完成,这些氨基酸标记被插入mAb的序列,能被酶识别(比如甲酰甘氨酸生成酶(formylglycine-generating enzyme FGE; also known as SUMF1), 转谷氨酰胺酶transglutaminases或 sortases)。SMARTag (developed by Redwood, now part of Catalent) 就是使用FGE技术的,tag为(Cys-X-Pro-X-Arg),识别这个tag后把半胱氨酸替换成甲酰甘氨酸formylglycine。修饰后的抗体能与乙醛特异的负载药物发生反应,这种反应是基于hydrazino-Pictet–Spengler ligation连接反应的。
Bacterial transglutaminases (BTGs) 也是此技术的一种。细菌分拣酶A(sortase A SrtA)是NBE Therapeutics开发出来的,这种酶是转肽反应的催化剂,这个酶识别的序列为Lys-Pro-Glu-Thr-Gly,当把这个序列置换入抗体重链和轻链的C端后,通过SrtA就可以把MMAE或者maytansine通过转肽反应偶联到五甘氨酸肽上。辉瑞采用这个技术用在I期临床试验管线PF-06664178上,靶点为TROP-2。Innate Pharma采用此技术把MMAE连接到IgG1上。
4.4 聚糖的改造和糖化聚合技术(Glycan remodelling and glycoconjugation)
天然的N-糖基化(Asn297)的IgGs是一种特异性位点修饰的技术,这个位点离抗原抗体结合去比较远,因此,可以最大程度上降低对绑定亲和力的影响,很多与N-聚糖(N-glycan)生物耦合的技术在研究中,包括代谢修饰(metabolic engineering),化学氧化(chemical oxidation),酶和化学酶的修饰。他们的共同特点是都能增加ADCs药物的同质性和稳定性。
4.5 高负载ADCs药物
高负载ADCs药物,通过传统的耦合,DAR为8,会有较短的药代动力学半衰期,更高的毒性,更低的治疗窗口。但随着位点特异性修饰技术的使用,高负载ADCs在到时模型中被证明其有效性,这是的临床上针对抗原低表达、低内吞、细胞内低效ADCs药物转运的肿瘤细胞被治疗成为可能。Mersana Therapeutics在研发聚合物为基础的ADC药物平台(Fleximers),使得每个抗体能携带15个负载,临床试验进展到了I期,由于试验期间有病人出现过死亡,所以相应管线被FDA暂停一段时间。
4.6 其他
氨基端修饰丝氨酸(Amino-terminal engineered serine);Fab核苷酸绑定连接技术(Ligation to Fab nucleotide-binding sites);天然半胱氨酸桥接技术(Native cysteine rebridging),这个技术有多家公司在研发,如PolyTherics (now Abzena), ThioLogics, Igenica Biotherapeutics, Sorrento Therapeutics and the University of Tours, France,用的是双烷基化连接抗体链之间的双硫键。这个技术的好处就是不需要再去特异性的对抗体的连接点进行修饰,从而不影响抗原抗体结合能力,并且能在血浆中保持稳定。避免Michael加成逆向反应的解偶联技术(Avoiding retro-Michael deconjugation),Michael加成用来把硫醇加入马来酰亚胺,在药物偶联抗体中经常使用,确实,在这两个FDA已批准的ADCs药物中都包含了马来酰亚胺-硫醇加成物,在体内试验中,这样的加成物通过硫醇交换反应被裂解,这样就会导致药效降低以及更高的系统性毒性。但是如果琥珀酰亚胺的马来酰亚胺-硫醇被水解,环状结构被打开,会生成结构更加稳定的产物,不少公司就在研发这种技术。有的为了避免马来酰亚胺耦合在循环中的不稳定性,使用有苯环的马来酰亚胺。
总结和展望
ADC药物的发展得益于抗体药设计的改进和特异性耦合技术的提高,多样化的连接技术和负载药物能明显增加药物的稳定性、同质性以及治疗窗口。蛋白结构鉴定工具(如质谱分析mass spectrometry)变得越来越重要,它能更好的识别ADCs药物在体内的多样化形态。这些知识的积累和越来越多的生物分析检测手段能对早期的药物研发的标准制定有重要作用。比如,肝细胞毒性的脱靶效应在不少ADCs药物中被报道,主要原因是因为肝脏细胞表面表达甘露糖受体,因此抗体分子中含有较低的甘露糖应该被优先选择。还有比如马来西安亚胺连接技术的替代技术,这类技术能很好的解决血液中非特异性释放的问题。
由于ADC药物是抗体药物和小分子药物的复合体,它的产品质量属性一定需要被很好的认识,因此,早期研发的药物评估就需要最先进的分析和结构方法,比如质谱分析(mass spectrometry)、二维液态色谱分析法(two-dimensional liquid chromatography)和毛细管电泳法(capillary electrophoresis),这些新方法能深入认识与ADCs功能相关的重要结构。
ADCs携带的小分子负载药物也是被关注的领域,未来需要在这类药物的机制上有所突破。ADCs中的抗体药物也有较好的替代物,比如,蛋白折叠结构相关的(DARPins, nanobodies, single-chain variable fragments (scFvs), peptide–drug conjugates),抗体双药偶联药物(antibody–dual-drug conjugates ADDCs),Fabs,Probodies (developed by CytomX)正在被研发当中。有意思的是第一个针对于双标位的(HER2靶点)的ADC药物已进入临床I期,部分数据已公布,此药可以和T-DM1在毒理、有效性、药代动力学上进行比较,以此可以看到能在治疗上到底带来多大的价值。
在治疗指征上,ADCs药物有比较多的选择可能,比如单药用于复发难治性肿瘤,保守姑息治疗,维持治疗,与其他药物联合作为一线用药或复发后的病人。一些随机对照研究也能更好的比较不同组合之间的毒性属性成本等。
全文完



