 下载格隆汇APP
下载格隆汇APP
 下载诊股宝App
下载诊股宝App
 下载汇路演APP
下载汇路演APP
 社区
社区
 会员
会员
微信公众号:蹊之美股生物医药 / CaesarBiotech
摘 要
抗体-药物偶联物 (Antibody–Drug Conjugates ADCs) 药物是肿瘤治疗领域内发展最快的一类药物,它具有抗体药的高选择性、稳定性、不错的药代动力学,同时具有小分子细胞毒性药物强力肿瘤杀伤作用,具有较大的治疗窗口。2011年西雅图基因公司/武田的ADC药物Adcetris和2013年罗氏旗下Genetch的ADC药物Kadcyla上市以及销售额的不断增长,证明了ADCs药物在临床中的意义和价值。ADCs研发技术上非常具有挑战,在经过半个世纪的研究,这一领域已积累了很多成功和失败的经验,但笔者认为这领域有可能成为肿瘤治疗的最有前景的治疗手段。
ADCs药物杀伤机制由多个环节组成:抗体-靶点结合,内吞作用,胞内药物或毒性基团的释放,药物在细胞器内的转运,药物到达靶点触发细胞凋亡,旁观者效应(Bystander Effect)等。此文主要讨论决定这些ADCs药物效能和安全性的关键技术,同时也会提及不少纳斯达克上市公司和非上市公司。
共五块内容(由于篇幅较长,分2部分发表)
背景
1. 靶点-抗体的选择和优化
2. 连接子的设计和优化
3. 细胞毒药物的选择及研发公司
4. 新型的特异性位点偶联技术
背景
恶性肿瘤是世界上死亡率排第二的疾病,目前对恶性肿瘤的主要治疗手段有:外科手术切除,化疗,放疗,肿瘤免疫治疗等。在这些治疗中,化疗可以单独使用治疗肿瘤,也可以与手术联合使用,或与放疗联合使用,是频率最高的治疗选择。新辅助化疗(Neoadjuvant)是在手术前或者放疗前联合化疗,以期缩小肿瘤。辅助化疗(adjuvant)是在手术后或者放疗后使用化疗,以杀灭可能残留的肿瘤细胞。传统的化疗药由于有较差的药代动力学,治疗窗口(therapeutic window)很低。而且,化疗药物并非特异性针对肿瘤细胞,对正常细胞也有毒性作用,因此脱靶效应(Off-Target)很明显。
而抗体-药物偶联物(Antibody–Drug Conjugates ADCs)药物可以弥补这些缺点。ADCs药物是由抗体(antibodies)通过连接子(linkers)结合细胞毒性药物(弹头 Payload/Warhead)组成。ADCs药物在循环中需要处于稳定状态,可以被认为是一种前药(prodrug),药物结合靶点后,内吞入细胞,通常在溶酶体释放细胞毒药物,进行肿瘤细胞杀伤作用。由于抗体靶向性强,携带负载药物的细胞毒性效能很高,次纳摩尔的半数最大抑制浓度(half-maximal inhibitory concentration IC50)就可以用来衡量负载药物的毒性了。下图是ADC药物的主要组成部分(见图下)。
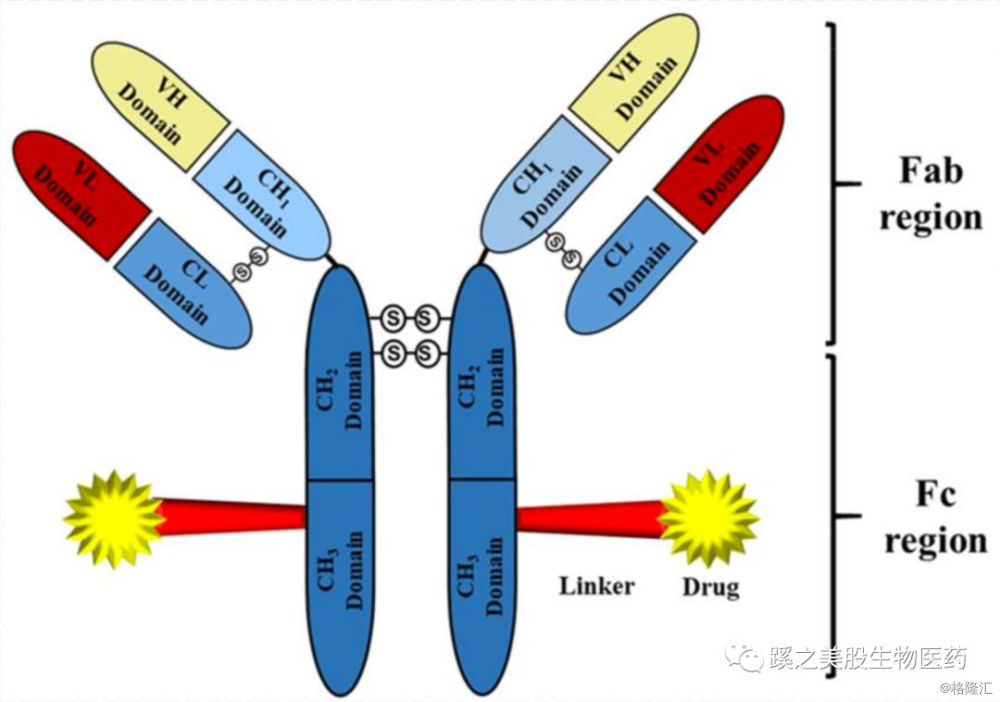
在上世纪60年代就有文献报道ADCs药物用于动物模型,到80年代就有鼠原抗体的ADCs药物进行临床试验,第一个ADCs药物是2000年批准的gemtuzumab ozogamicin GO (developed by Wyeth),靶点为CD33,负载药物为卡里奇霉素(Calicheamicin),用来治疗急性髓样淋巴瘤(acute myeloid leukaemia AML),然而在随后的批准后研究中发现GO联合化疗药不能有效提高生存时间,而且相对于单用化疗有更高的致命毒性,在2010年,由辉瑞(辉瑞收购了Wyeth)自行撤离市场,此药没有在欧洲批准。随后,2011年Seattle Genetics/武田的Adcetris (brentuximab vedotin 靶向CD30)和2013年罗氏旗下Genetch的Kadcyla(trastuzumab emtansine 靶向HER2)被美国FDA及欧洲EMA批准上市。之后,ADCs药物的研发不愠不火,大概有30多个ADCs药物进入临床试验。
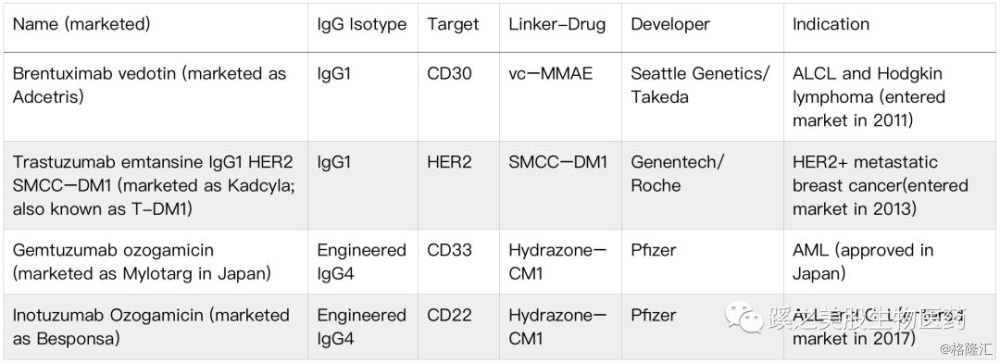
至今已有4个ADCs药物被美国FDA及欧洲EMA批准,还有个治疗三阴性乳腺癌(TNBC)药物sacituzumab govitecan(由Immunomedics公司研发),临床数据很不错,但由于CMC的问题,目前尚未被FDA批准。目前有超过70个ADCs候选药物进行临床试验。第一代、第二代、第三代ADCs药物不断的技术迭代,更同质性、更稳定、杀伤效能更高的药物研发不断出现。已被批准的ADCs药物见下表(表格1)。
表格1 已被批准的四个ADCs药物(作者整理):
负载药物中,有将近2/3集中在奥瑞斯他汀(auristatins)和美橙木素生物碱(maytansinoids)两大抗有丝分裂药物家族上,负载药物需要高效能、相对的亲水性、不能被多重耐药蛋白1(multidrug resistance protein 1 MDR1)介导的泵排出,同时还需要有合适的化学键能完成与抗体及连接子的连接,不少临床试验已经失败过。
连接子的优化对于ADCs药物的研发非常重要,当ADCs药物在循环中时,连接子必须十分稳定,以保证最小的脱靶效应(off-target)产生,在进入细胞内部时,释放效率需很高。最近的一些研究表明,大部分二代ADCs药物在循环中的稳定性其实也是有限的,大部分二代ADCs药物用的马来酸亚胺类型的连接子,在血浆中会出现早期解偶联现象,这会导致一定的脱靶毒性。这种现象在药物连接至抗体的赖氨酸及半胱氨酸残基上尤为明显。在第三代ADCs药物的研发中,有更好的技术来解决这个问题,下文将会有所讨论。
另一个主要的技术是,在IgG分子的特定位点上进行修饰,用以连接细胞毒药物,这样可以使产生的ADCs有更好的同质性和稳定性。最初,有人把IgG1铰链区的8个天然丝氨酸中的2个或4个突变为半胱氨酸,这些在突变替代的轻链和重链位置的半胱氨酸能提供硫醇基团,用来提供连接的残基,当然也可以在其他地方做突变替代。这种技术叫TDCs(THIOMAB drug conjugates TDCs),这样可以生成接近一致的药物抗体比(Drug‐to‐Antibody Ratio DAR)为2或者4的ADC药物,而不用破坏抗体上的二硫键(半胱氨酸桥),不幸的是,第一个TDCs药物在循环中也存在较高的解偶联率,这是由于马来酸亚胺(maleimide)可以与血浆中的白蛋白、自由半胱氨酸、谷胱甘肽发生硫醇基团的交换反应。这个问题在下一代TDCs药物的研发中得到解决,当把特异性位点选择在阳性电荷环境中时,可以导致琥珀酰亚胺的环被水解,从而防止硫醇基团的交换反应。
ADCs药物在循环中的不稳定性,可以通过特异性位点偶联技术(site-specific conjugation)得到解决,目前有超过40个这样的技术在研发,并研发相应的偶联化学反应技术,至少10个使用这样技术的ADCs药物进入临床试验。这些技术的目标都是都是用来增强药物的同质性,减少药物在循环中的释放。下面,我们就来讨论下靶点的选择、抗体的优化和选择、连接子的设计和优化,细胞毒药物的种类及特点,特异性位点偶联相关技术等。
1. 靶点-抗体的选择和优化(Target and Antibody)
靶点和抗体结合的特异性直接决定了细胞毒药物能否精确作用于肿瘤细胞,保证有效性的同时,降低脱靶效应。因此,决定什么抗原作为靶点是研发ADC药物重要的一步。免疫组化、流式细胞仪、组织基因测序、RT-PCR、mRNA等技术通常被用来分析病人样本中肿瘤细胞表面表达的抗原。在确定表面抗原过表达后,通过杂交瘤技术(Hybridoma Technology)来制作抗原特异性的单抗。杂交瘤细胞是通过融合小鼠的抗体产生B细胞和骨髓瘤细胞产生的永生化细胞。然后反复传代来生产足够的抗体。根据抗体在肿瘤细胞中的渗透能力和结合亲和力(Kd < 10 nM),来选择做为ADC药物的抗体结构。抗体亲和力强的会局限在血管壁周围空间里,而低亲和力抗体能较好的渗透至肿瘤细胞之间,被内吞至肿瘤细胞内。因此,掌握内吞和抗原抗体复合物分解率的平衡,能决定细胞毒药物递送至肿瘤的效率。
1.1 靶点抗原的选择
1.1.1 首先,为了减少脱靶毒性和产生可以接受的治疗参数,理想的靶点抗原应该在肿瘤细胞表面高表达,在正常组织相对低或者没有表达。在不少临床试验进行中的管线中,有些靶点只对某种肿瘤具有特异性,有些靶点可以针对多种实体肿瘤类型,(比如,5T4 又称为trophoblast glycoprotein,在多个实体瘤表达;胰腺癌和卵巢癌的靶点间皮素mesothelin;CD138,又称为SYND1,在多发性骨髓瘤和不少实体瘤中表达)。在血液病恶性肿瘤中,如靶点SAIL(surface antigen in leukaemia)和CD37,它们在这类肿瘤中广泛表达。还有ADCs药物可以设计成针对于肿瘤微环境靶点,比如,tetraspanin-like protein transmembrane 4 L6 family member 1 (TM4SF1) ,能同时表达于肿瘤细胞和肿瘤脉管系统。甚至,一个三功能的抗体-细胞因子-药物偶联体能作用于肿瘤微环境中的纤连蛋白(fibronectin)。
1.1.2 其次,靶抗原应该在细胞表面表达,这样能被循环中的抗体结合。
1.1.3 再次,抗体和抗原结合后应该能被细胞内吞,这样ADC药物能转至细胞内发生作用。但是,也有报道表明,非内吞的ADC药物也能表现出不错的细胞毒性作用,这些ADC药物往往通过较强的“旁观者效应(bystander effect)”来起到杀伤作用。这些靶抗原
相对应的裸抗体不是一定要有细胞杀伤作用。在HER2靶点的ADC药物研发过程中,首先是mAb的trastuzumab被罗氏研发出来,此抗体被用来研发ADC药物(trastuzumab emtansine T-DM1),细胞毒药物用的是美橙素衍生物DM1(N2'-deacetyl-N2'- (3-mercapto-1-oxopropyl)-maytansine)。T-DM1具有细胞外肿瘤杀伤机制,比如ADCC和ADCP(antibody-dependent cell-mediated phagocytosis)。与此不同的是,被FDA批准的另一ADC药物brentuximab vedotin,作用于多种淋巴细胞增殖的疾病,这是CD30抗体,只显示针对间变性大细胞淋巴瘤(anaplastic large-cell lymphoma ALCL) 肿瘤有中等细胞外肿瘤杀伤机制,其血液肿瘤细胞则没有。在抗CD38抗体的研发中也有类似的发现。因此裸单抗的抗肿瘤活性对研发ADC药物不是一定需要的。ADC药物的设计中,需要综合考虑细胞毒药物的毒性,抗体药物的抗肿瘤作用,以及整个ADC药物的细胞毒作用特点。
有些裸抗靶点已经上市了的,比如B细胞非霍奇金淋巴瘤(B cell non-Hodgkin lymphoma B-NHL)的CD20抗体,还有CD19, CD22, CD79b等。有些裸抗的靶点还没上市,比如三阴性乳腺癌(triple-negative breast cancer TNBC)靶点,辉瑞的PF-06647263的ADC药物,靶点为ephrin A4,细胞毒药物为卡里奇霉素类,在临床前试验中针对TNBC和卵巢癌获得不错的结果,Celldex 的Glembatumumab vedotin (also known as CDX-011) 针对于TNBC。
1.2 ADC药物抗体的选择和优化
增加抗体的同源性和可研发性对裸抗体和ADC药物的研发都很重要,流式细胞仪、电泳技术、光谱分析被用在了抗体的研发、临床前和临床试验中。这些分析可以筛选更好的糖基化特征、稳定性高的、药代动力学好的克隆,可以研发出CMC成本低、成药性更好的抗体药和ADCs药物。
1.2.1 嵌合抗体、人源化抗体、全人抗体 在命名上,WHO有一套针对抗体药物的国际通用命名方法,比如“-Mab”后缀的一般都指单克隆抗体,“xi-”词根的通常为嵌合抗体,“zu-”通常指人源化抗体,或者“u-”指全人抗体,这样的命名可以把抗体的免疫原性考虑进去。但WHO最近开始根据氨基酸序列来修改命名法,这可能会使一些命名产生混淆,所以,在实际使用上,需要加以鉴别。
1.2.2 抗体类型及结构特点 大部分批准的抗体药,都来自3个IgG亚型(IgG1, IgG2 or IgG4),IgG3半衰期低,相对其他类型有扩展的铰链区域,有多态性和免疫原性。亚型分类是通过抗体中的重链氨基酸序列来区分的。二硫键(IgG1和IgG4有16个,IgG2有18个)和非共价作用维持抗体的三维结构,重链和轻链通过1个二硫键连接,重链之间通过2个(IgG1和IgG4)或者4个(IgG2)双硫键连接,这些双链在高度可变的铰链区域(hinge region)。其他12个双硫键在抗体分子内部,界定着抗体的6个不同球状区域,1个位于轻链的可变区(variable VL),1个位于轻链的恒定区(constant CL),1个位于重链的可变区( variable VH),另外3个分别位于重链剩下的三个区域(CH1、CH2、CH3)。简单来说,抗体的每个区域内部都有一个双硫键(结构见第一张ADC药物的示意图)。
不管是裸抗体或者ADCs上的抗体药(包括已上市的brentuximab vedotin,tras- tuzumab emtansine),抗体的类型多为IgG1亚型(包括嵌合抗体、人源化抗体、全人抗体),IgG1在制作ADCs药物时要相对于IgG2容易,因为IgG2有着不同的同分异构的双硫键连接着更加复杂的铰链区域。抗体的Fcγ区域,能触发免疫介导的效应功能(Effector Function),比如抗体依赖的细胞毒作用ADCC(antibody-dependent cellular cytoxicity)和补体依赖的细胞毒作用CDC(complement-dependent cytotoxicity)。不同的IgG亚型,它们的这种免疫功能是不同的,IgG1通常都具有ADCC及CDC功能,IgG2和IgG4相对较弱,IgG4可能具有ADCP(antibody-dependent cell-mediated phagocytosis)功能。
抗体的细胞毒作用能增强ADC药物的细胞杀伤作用,比如HER2靶点的抗体赫赛汀trastuzumab,被制作成ADC药物T-DM1(trastuzumab emtansine)后,trastuzumab的ADCC作用被保留,绑定HER2靶点,防止Her2的脱落,并抑制phosphoinositide 3-kinase (PI3K)–AKT信号通路,抗体的Fcγ receptor (FcγR)起到了免疫细胞的ADCC作用。T-DM1对拉帕替尼lapatinib(小分子Her2和EGFR通路抑制剂)耐药的肿瘤同样有作用。因此T-DM1的指征为治疗HER2阳性的,之前接受过trastuzumab和一种紫杉烷治疗的转移性乳腺癌病人。当然,也有公司认为ADCC再加上细胞毒药物可能毒性太高,如ADC药物 MEDI4276(developed by AstraZeneca/MedImmune 目前在临床I期),它的Fc区域(E234F, S239C and S442C)被突变掉,用以减少FcγR绑定以及减少低血小板症。
2. 连接子(Linker)的设计和优化
连接子是用来把细胞毒性药物连接至抗体的化学分子,它的物理化学特点很大程度上决定了一个ADC药物的药代动力学。有效的连接子能平衡循环中药物的稳定性和在目标细胞中的有效释放。设计上有多种策略能加强药物的可溶性和DAR指数,用来克服转运蛋白对化疗药物产生的耐药性,比如MDR1。这些策略包括有条件的在细胞液中释放负载药物,增强旁观者效应(通过非极化连接子-药物代谢产物,使细胞毒药物代谢物能穿透生物膜),限制旁观者效应(通过带电荷的连接子-药物代谢产物,使细胞毒药物代谢物不能穿透生物膜)。
连接子分为可分解(cleavable)和不可分解(non-cleavable)两种。连接子在设计上要求能使ADC药物在循环中很稳定,不释放药物,一旦到达细胞内的靶点,就会释放细胞毒性药物。这两种连接子都被用在第二代和第三代ADC药物的研发上。
2.1. 可分解连接子(Cleavable Linkers)
2.1.1 酸性敏感连接子 酸性敏感腙基团(化学结构见下图),连接子在循环(pH7.5)中稳定,到了溶酶体(pH4.8)和内吞体(pH5-6)后在酸性的微环境中分解。这些连接子是非特异性(nonspecific)的释放。2010年被撤出市场的gemtuzumab ozogamicin(developer Pfizer)就是用这种连接子。milatuzumab doxorubicin(also known as IMMU-115; developed by Immunomedics,stopped)和inotuzumab ozogamicin(developer Pfizer,临床前)中的连接子就能被低pH值环境水解)。但由于这类连接子可能在循环中有非特异性释放,所以不少临床试验因此而停止了。
2.1.2 谷光苷肽敏感性二硫键 它能被谷胱甘肽还原,在肿瘤细胞内因为氧化应激,硫醇基团明显增多,从而导致肿瘤细胞内和细胞外谷胱甘肽的浓度明显不同,这样ADCs药物进入细胞内时,双硫键就能断裂,除了谷胱甘肽,细胞内的蛋白双硫键异构酶(protein disulfide isomerase PDI)也能分解双硫键。美登木素生物碱药物广泛的使用这种连接子,平均的DAR在3-4左右。空间结构的双硫键桥能限制药物在细胞内被提早释放,这些双硫键起初被裂解成硫化的DM4,然后再在细胞甲基转移酶的作用下S-甲基化,才能起作用。比如ADC药物lorvotuzumab mertansine(developer ImmunoGen), coltuximab ravtansine(developer ImmunoGen) 和 anetumab ravtansine(developer Bayer)。

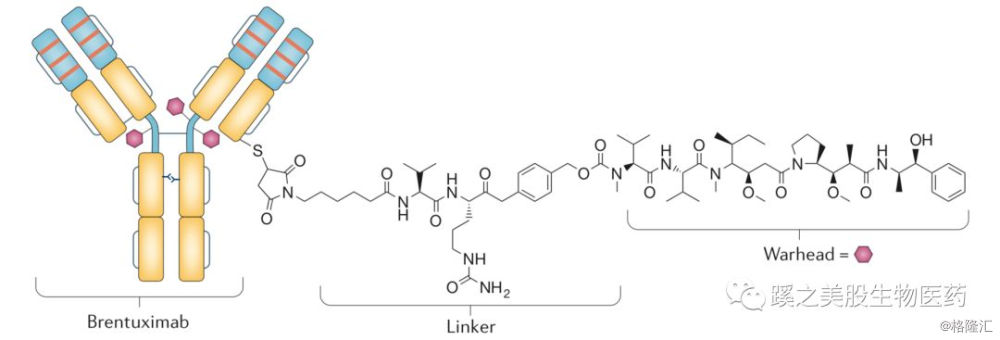
2.1.3 溶酶体蛋白酶敏感的肽连接子 肿瘤细胞中的溶酶体能表达高于正常的cathepsin B蛋白酶,这些蛋白酶在循环中由于高pH值和蛋白酶抑制剂,是没有活性的。这使得这种连接子在循环中能很稳定的存在,只在进入细胞内溶酶体内,连接子才能被分解,被FDA批准的Adecetris®(brentuximab vedotin developer Seattle Genetics/Takeda)的连接子就是cathepsinB敏感的缬氨酸-瓜氨酸连接子(Valine-Citrulline)。Adecetris®在后面被批准用于多个适应症,而且都是1线用药,证明此ADC药物的各个属性非常不错。(Adecetris结构见下图,Warhead为monomethyl auristatin E MMAE)
2.1.4 β-葡糖苷酸连接子(β-Glucuronidase-sensitive linkers) β-葡萄糖醛酸酶敏感的连接子已被用于不少葡糖苷酸的前药,溶酶体和肿瘤坏死区域富含β-葡萄糖醛酸酶,这种酶在溶酶体酸性环境中具有活性,而在生理状态下的pH值没有活性。β-葡萄糖醛酸酶能把β-葡萄糖醛酸酶敏感的β-葡糖苷酸的糖苷键分解。而且这种连接子有亲水性,能用于疏水性的负载药物,用来降低ADCs药物的凝聚。
2.2. 不可分解连接子 (Non-Cleavable Linkers)
相对于可分解连接子,含有不可分解的硫醚连接子的ADCs在血浆中有更好的稳定性,非特异性的药物释放也较少。连接子通过非还原化学键连接至抗体的氨基酸残基上,内吞后,ADCs药物在溶酶体被降解成药物-连接子-氨基酸残基(drug-linker-amino acid residue)发挥杀伤作用。被批准的药物trastuzumab emtansine (Kadcyla®/T-DM1),使用的是不可分解的SMCC(N-succinimidyl-4-(maleimidomethyl) cyclohexane-1-carboxylate)连接子(见下图左),在溶酶体被裂解成lysine-MCC–DM1具有细胞杀伤作用,lysine-MCC–DM1由于疏水性不能作用于肿瘤旁的细胞,因此没有旁观者效应。当DM1使用四聚体PEG4Mal(见下图右)连接子时,极性增加,对于MDR1阳性的肿瘤细胞,杀伤力更好。再如,马来酰亚胺基己酸(maleimidocaproic acid)连接子(mc–MMAF),被用在depatuxizumab mafodotin上,最后被代谢成cysteine-mc–MMAF而发挥作用,也被证明有更高的效能,循环中的稳定性更高。
2.3 调控旁观者效应在ADC药物中发挥的作用
实体瘤能表达异质性的抗原,ADCs药物能选择性的杀灭抗体阳性的肿瘤细胞,同时对周围抗原阴性的肿瘤细胞不能起到很好的杀伤作用,因此ADCs药物也许可以设计成不仅能杀死抗原阳性的肿瘤细胞,同时不管周围的肿瘤细胞抗原表达是否阳性,也能杀灭,这就是“旁观者效应(bystander effect)”。先简单看下ADC药物内吞及细胞内杀伤机制。ADC药物胞转运过程经典模型及旁观者效应,见下图。

从图中可以看到,ADC药物先与细胞膜上的抗体相结合,然后抗体抗原复合物通过细胞内吞作用进入细胞内,并形成内含体,随后内含体进一步成熟并融合成溶酶体,在溶酶体内,细胞毒药物被释放(通过与之相对应的特异性蛋白酶分解连接子,如组织蛋白酶B(cathepsin B),或者整个ADC药物在溶酶体被分解),产生的细胞毒药物穿透溶酶体膜,绑定至DNA或者微管导致细胞凋亡。这些药物也可以通过细胞膜上的蛋白转运泵出至肿瘤微环境,进入到相邻的肿瘤细胞上,对其产生杀伤作用,这就导致了“旁观者效应(Bystander Effect)”。相邻的肿瘤细胞可以为细胞表面抗原阴性的肿瘤细胞,这样旁观者效应就会对抗原阴性的肿瘤也产生杀伤作用。这种旁观者效应在设计ADC药物时,也越来越多的被考虑,因为这只要求肿瘤中一部分细胞携带目标抗原即可达到对抗原表达阴性肿瘤细胞的杀伤作用。
比如像brentuximab vedotin(marketed as Adcetris, developer Seattle Genetics),被裂解后释放MMAE,MMAE能够穿过细胞膜杀伤相邻的上皮细胞。与之相反的,denintuzumab mafodotin(developer Seattle Genetics),连接的细胞毒药物为MMAF,裂解后的细胞毒药物代谢物有羧基端的苯基丙氨酸残留,这种代谢后的产物不能穿透生物膜,因此,相对于MMAE产生很少的旁观者效应。所以虽然单独的MMAF的细胞毒性比MMAE要强,但做成ADC药物后,由于有旁观者效应,ADC药物的实际细胞毒性会变得有所不同。同样的,可还原的双硫键有旁观者细胞毒效应,而非还原性硫醚连接子没有旁观者细胞毒效应(比如trastuzumab emtansine)。
肿瘤细胞能通过上调MDR1的表达产生耐药性,美橙木素为基础的ADC药物,由于不带电荷,或者说非极性连接子,对于MDR1阳性的肿瘤细胞仅有很低的杀伤力。MDR1对与输水化合物相对于亲水化合物有更高的转运效率。因此,带电荷的或者亲水的连接子正在被研发,可以增强对MDR1阳性细胞的杀伤效率。ImmunoGen的Mirvetuximab soravtansine (also known as IMGN853)对于铂类耐受的卵巢癌有比较好的效果。



